Computational Chemistry, as a Physicist
The moment I got into SUNY Binghamton, I knew I would need something more than classes to satisfy my curiosity. From the first semester I looked for research groups with work that interested me. It wasn’t until I met the faculty of Chemistry that I found a group who’s work fascinated me. After a semester of training, I started working with Prof. Dr. Puja Goyal at my second year at Uni. My first real research position!
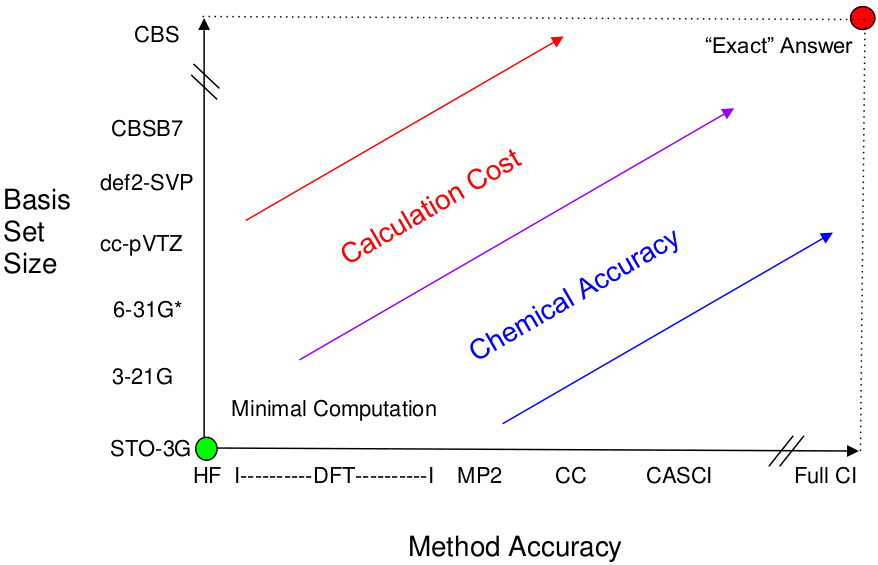
The Computational Space for Electronic Structure Calculations
From my Bachelors’ Thesis
I worked with the Goyal computational chemistry group for 3 years, sophomore to senior, and also wrote my senior thesis in the group. In those 3 years I worked on a variety of projects, using atomistic molecular dynamics and quantum chemistry calculations to study chemical and biochemical molecules. I wrote my final thesis on using different methods to reproduce UV-visible spectra.
The projects I remember best were ones that connected the chemistry to physics.
I projected the high dimensional conformational space of a protein into its principle components. I remember vividly, being shocked that molecular machines were in fact very flexible, while remaining functional.
I used molecular dynamics to study the see if a photoreceptor could control a signalling protein. Specifically, I studied the photocontrol of Calmodulin. The idea that theoretical work like this, would directly be useful to scientists using light to control matter was astonishing.
But by far, the aspect that excited me the most was that the physical quantum descriptions of the molecules, influenced the biological scale, and that we could actually calculate this.
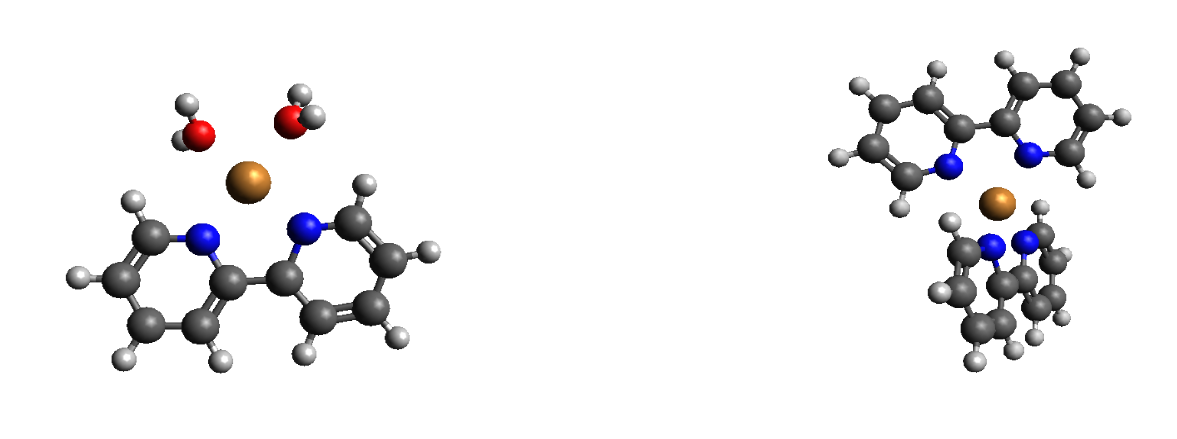
From my Bachelors’ Thesis, Optimized geometries of two copper complexes. The left complex has metallic oxidation states of Copper(1) and the right Copper(2)
I mainly studied two types of systems, Organometallic Copper complexes (shown above) and Azobenzene photodyes (discussed below). Starting with density functional theory (DFT), I progressed to calculating absorbance spectra using linear response theory, also known as time dependent DFT (TD-DFT). Since I had a bit of experience with various methods, we decided to focus on comparing said methods. I was so happy to see the things I did for research being mentioned in my quantum mechanics class. Near the end I used state of the art methods, like floating occupation molecular orbital complete active space configuration interaction (FOMO-CAS CI), and cheap and dirty approximation such as density functional tight binding (DFTB). I realized that a part of the job as a computationalist is to find the cheapest method that still gets the job done.
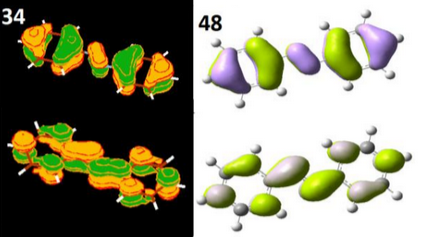
Quantum Calculations of cis- and trans- Azobenzene
From my Bachelors’ Thesis
Top Left: Comparing the highest occupied and lowest unoccupied molecular orbitals with DFT on the left and FOMO-CAS CI on the right
Top RIght: Structures of cis- and trans- azobenzene after geometry optimization using DFT and DFTB
Bottom: Absorbance spectra for cis- and trans- azobenzene from TD-DFT and TD-DFTB calulations
My time as a computational chemist left a huge impact on my trajectory as a scientist. It was a perfect middle ground, when I was torn between the rigid view of nature taught in my physics classes, and my childhood inclination for biological systems.